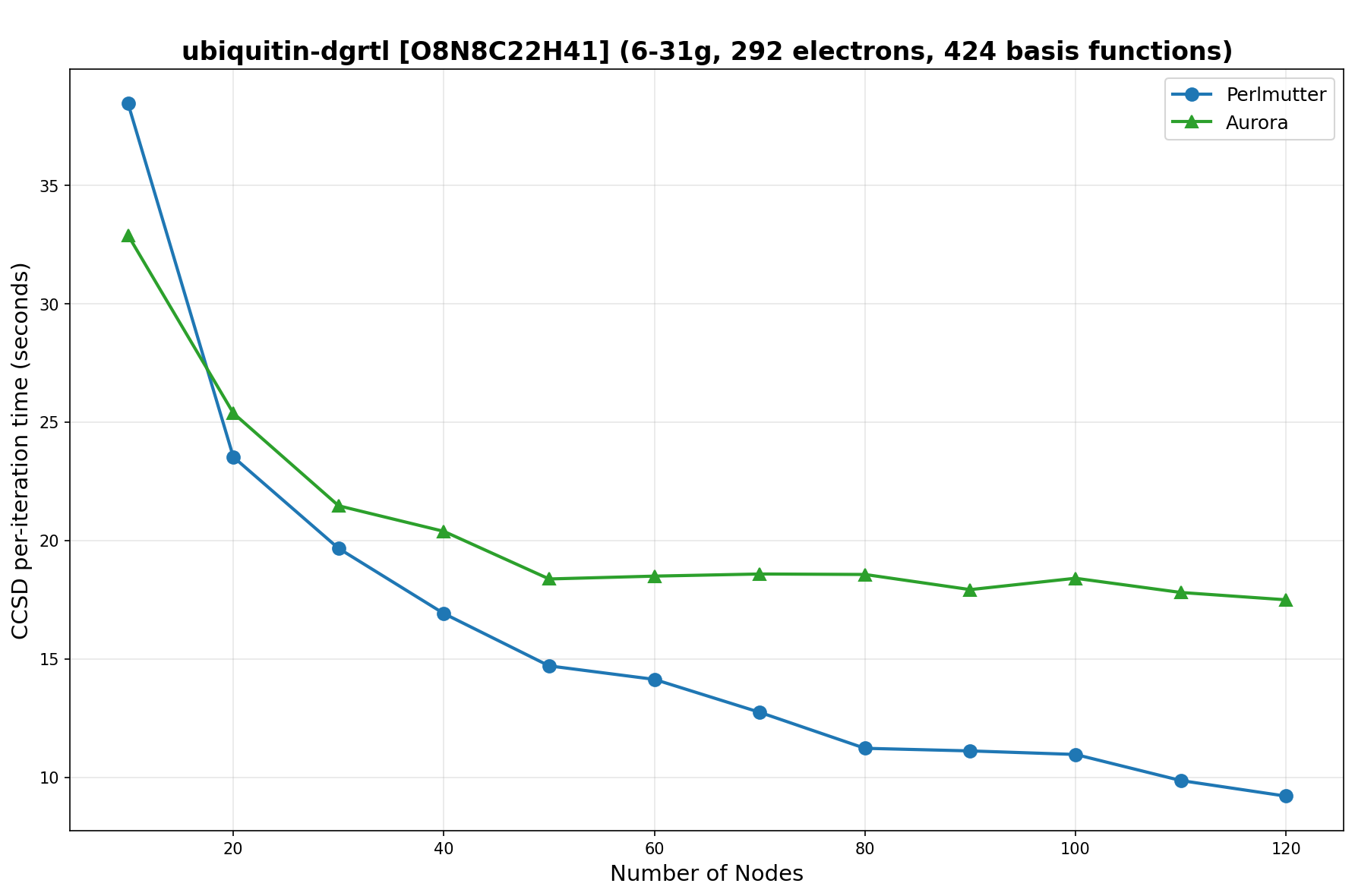
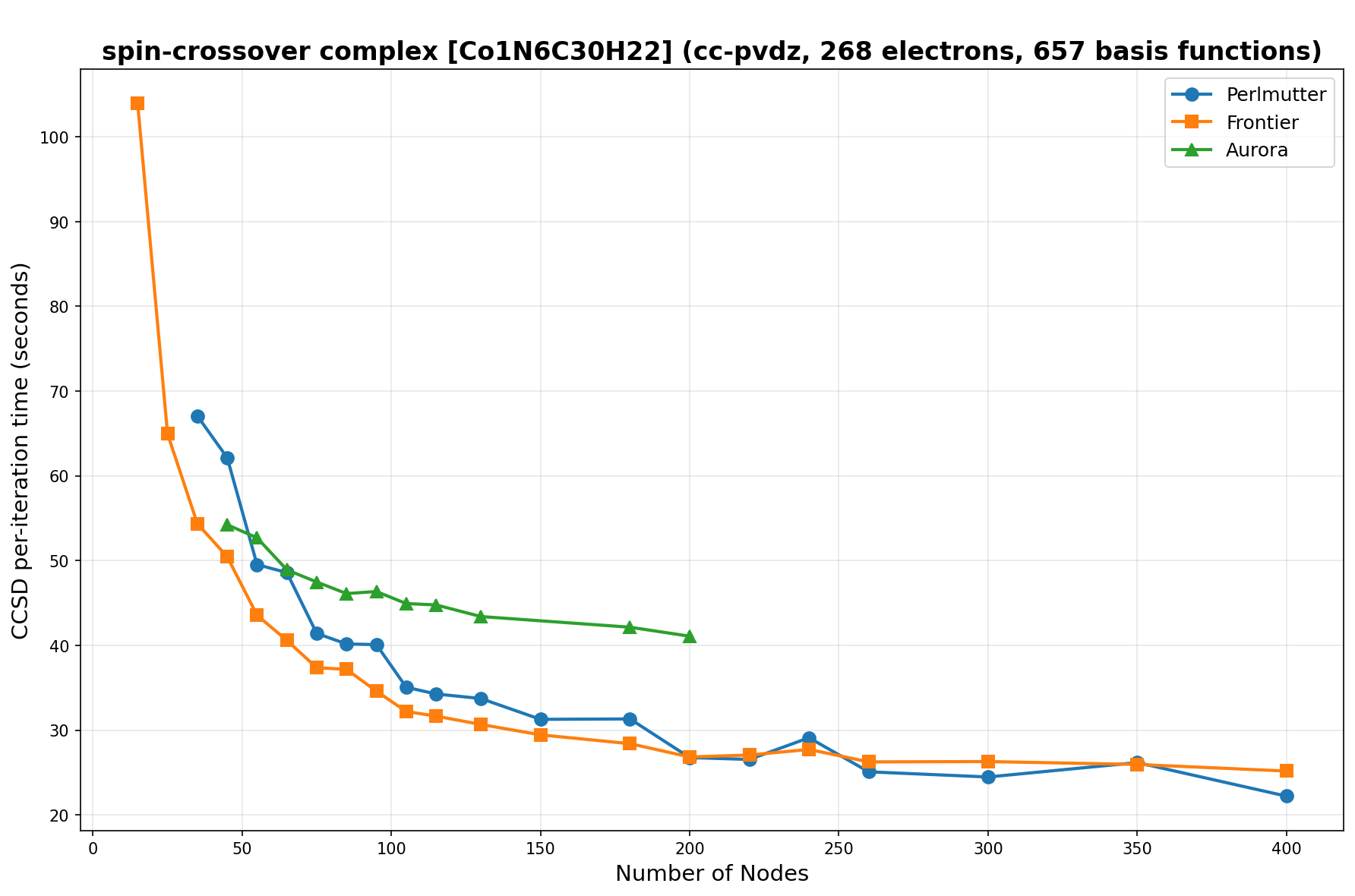
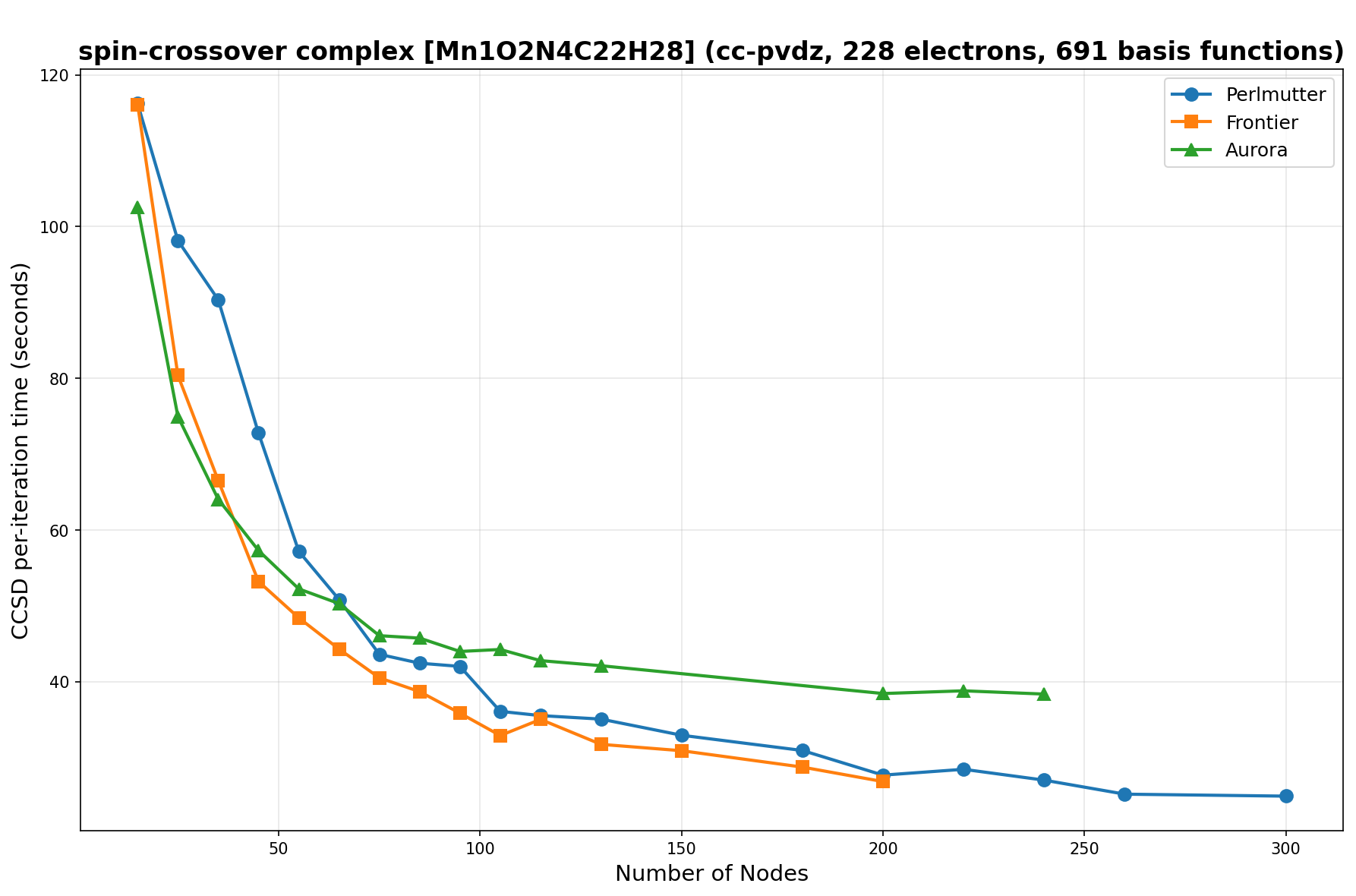
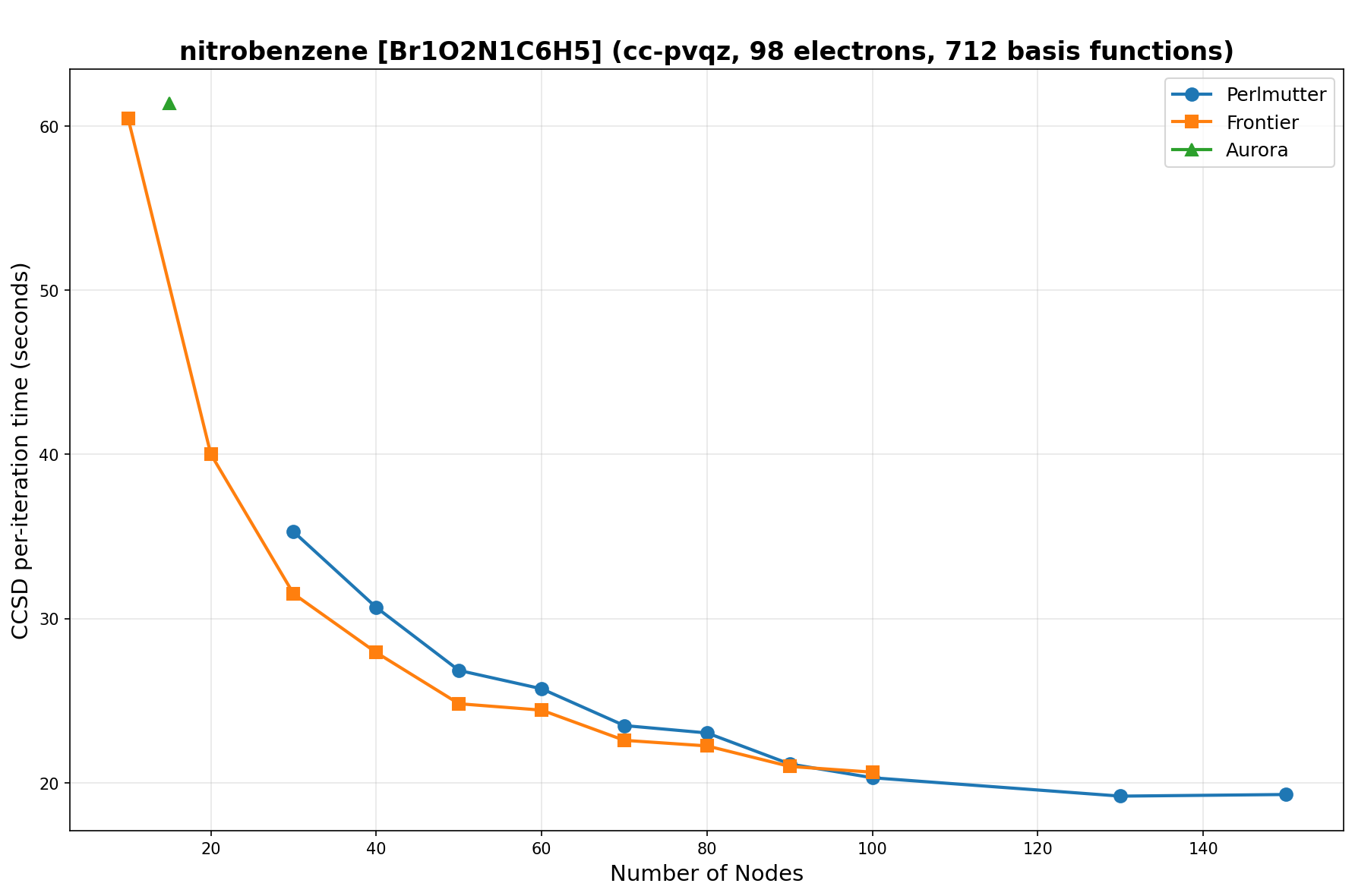
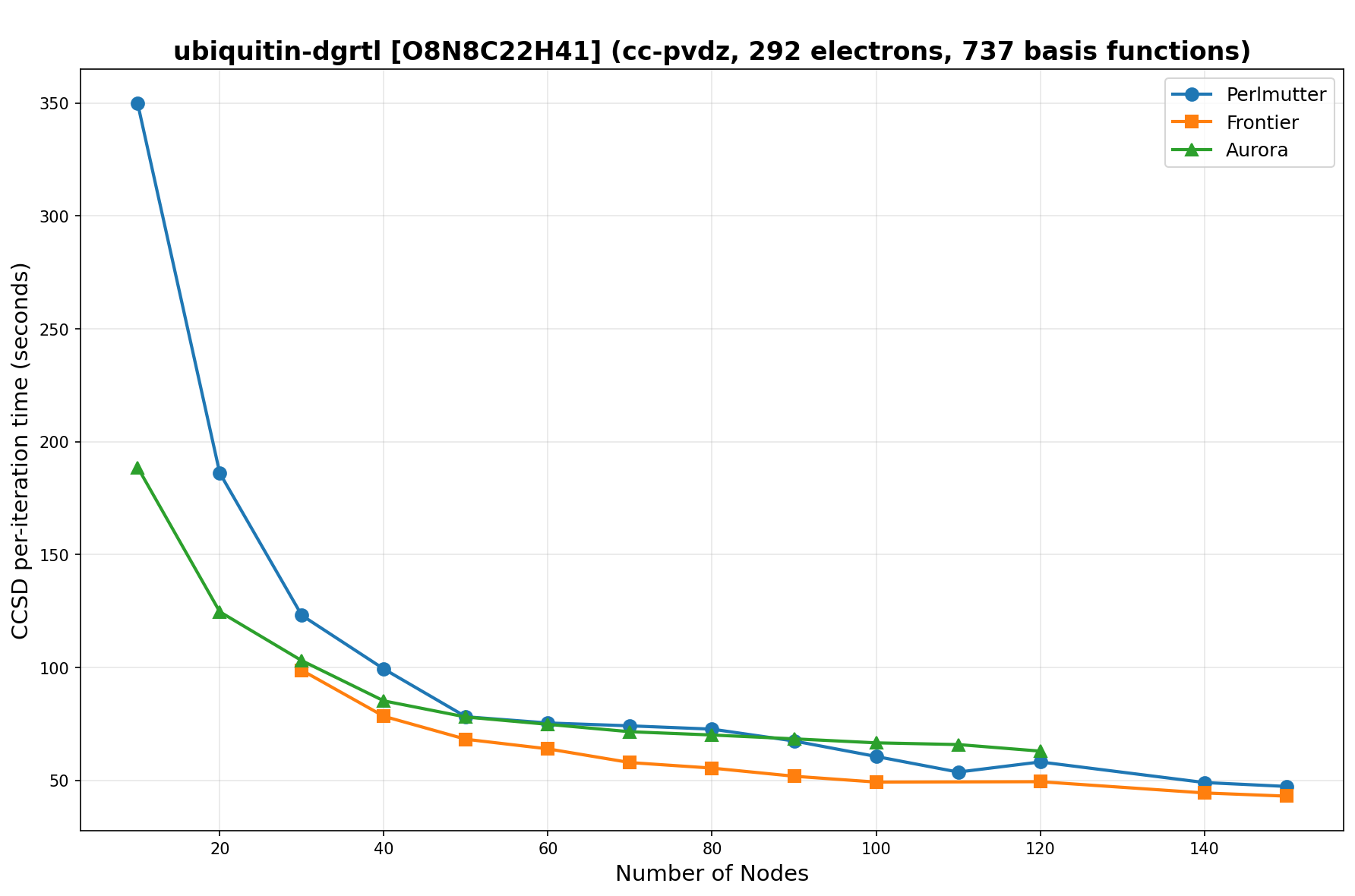
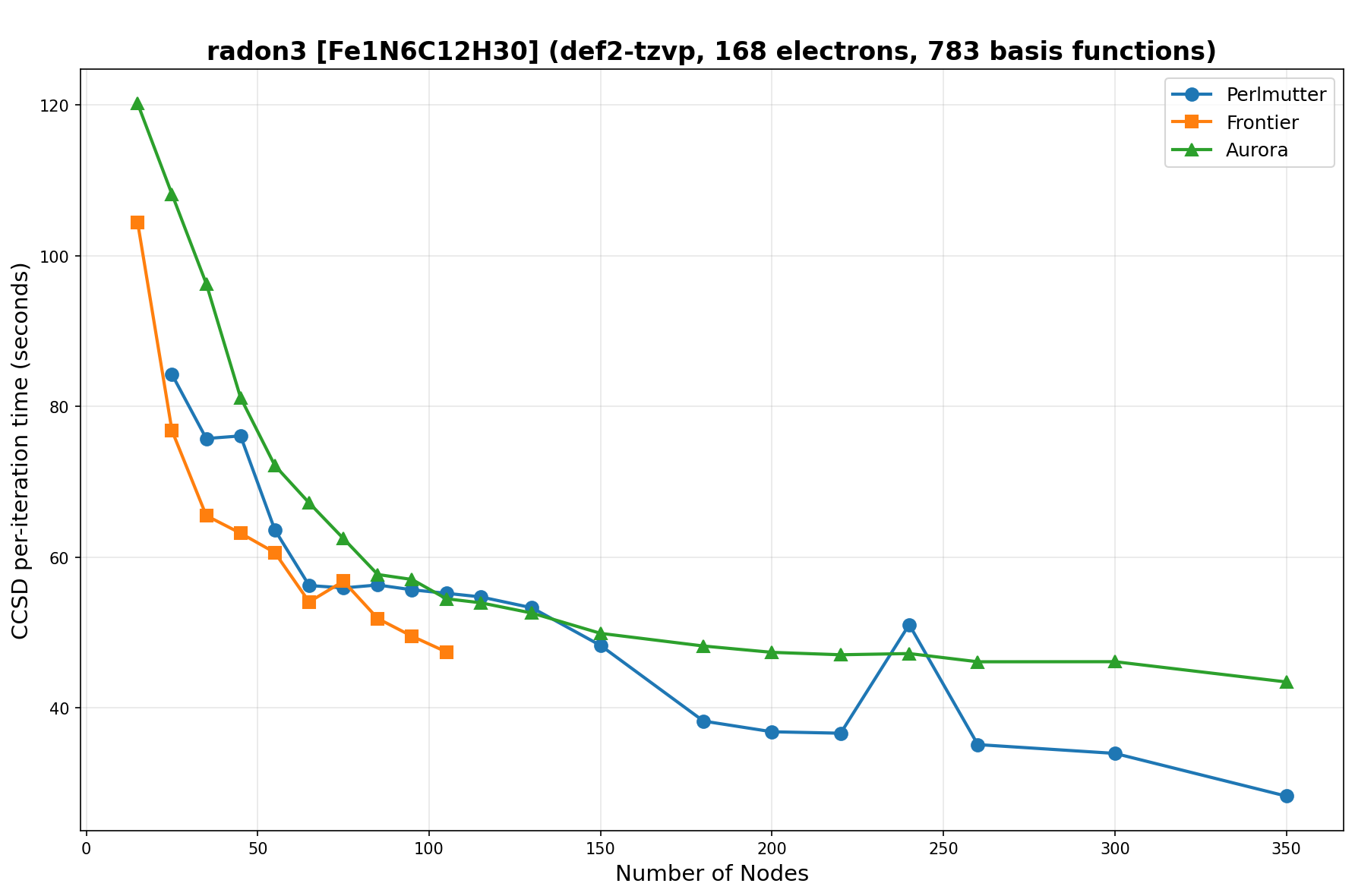
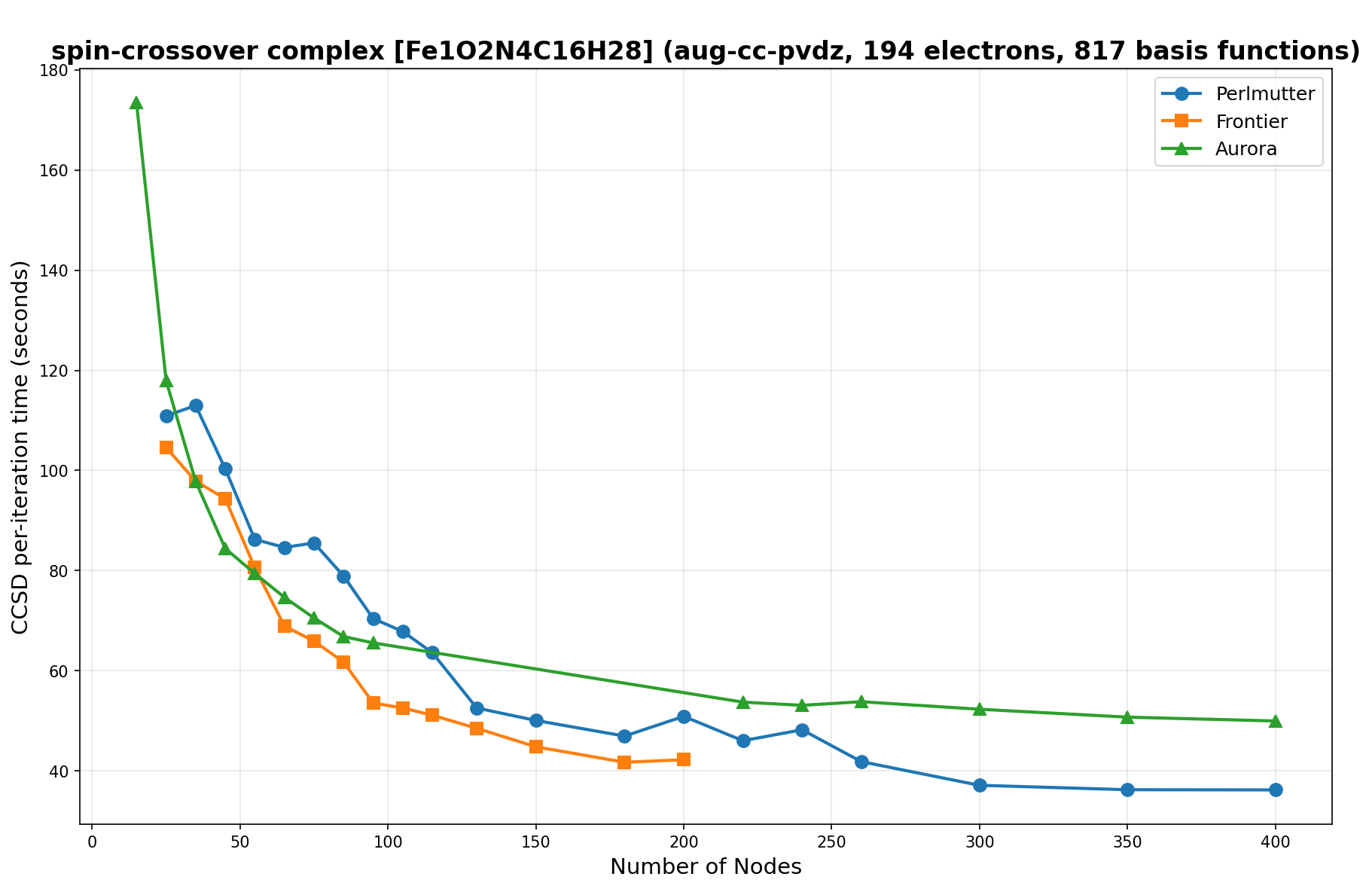
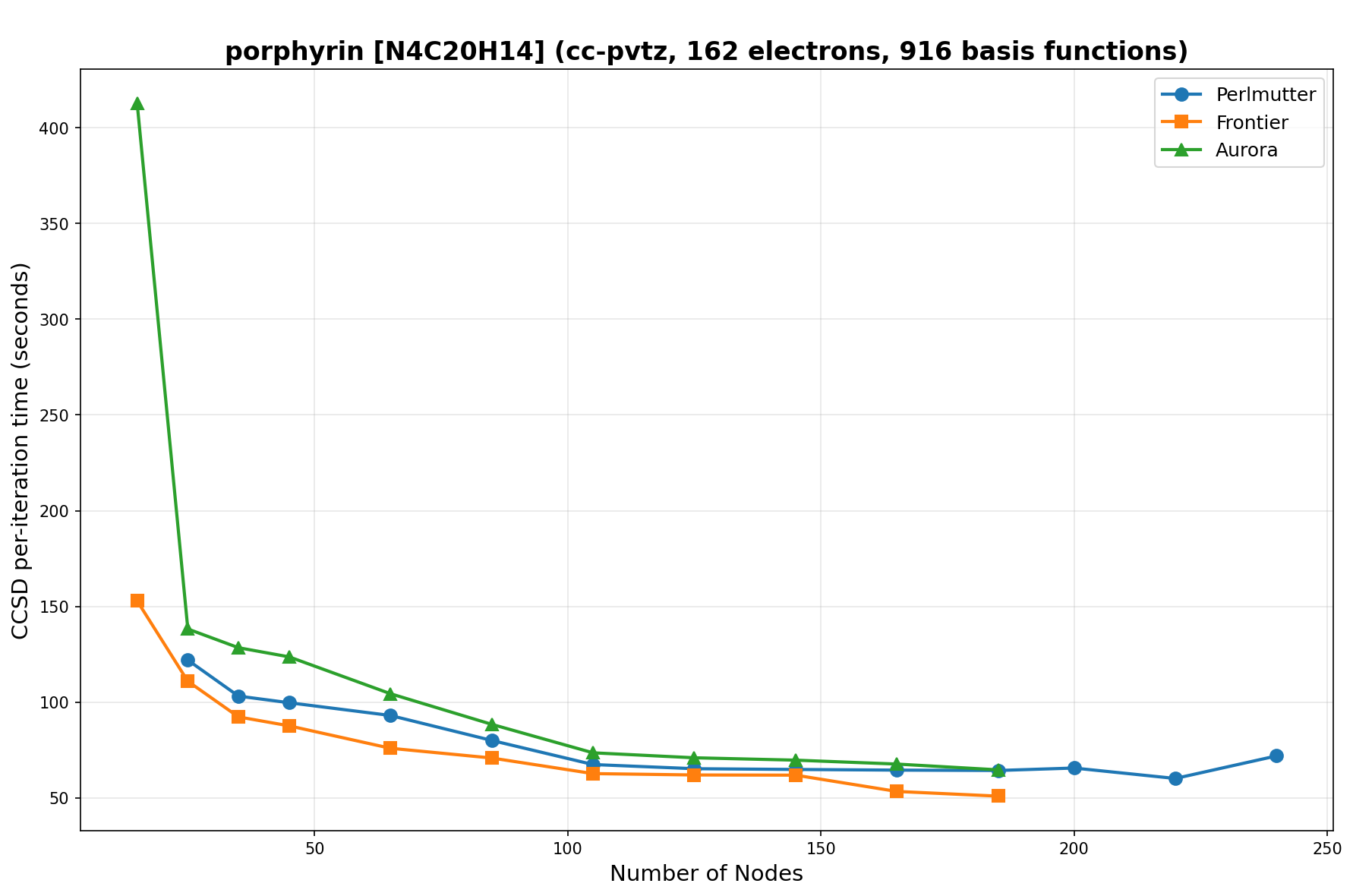
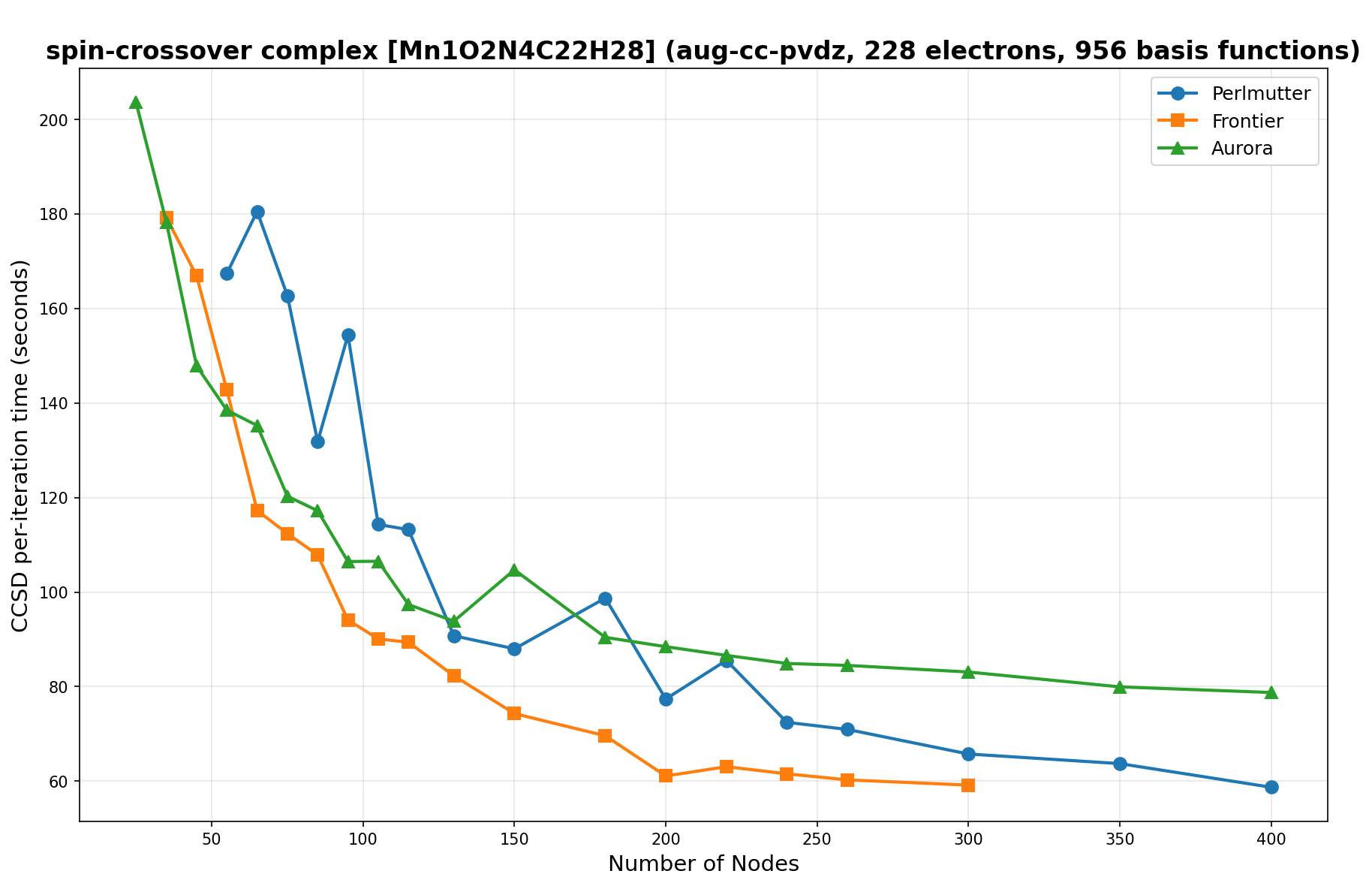
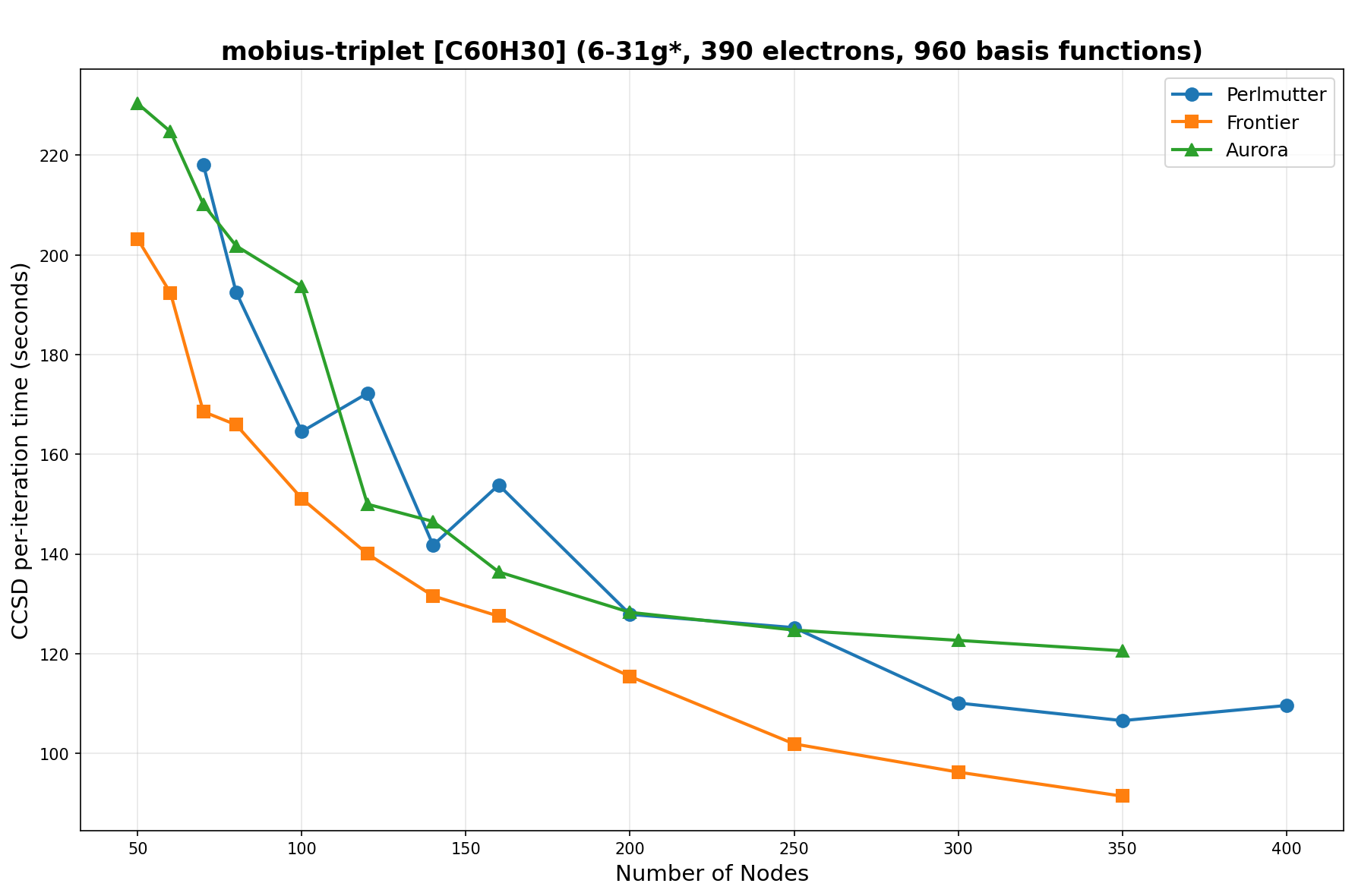
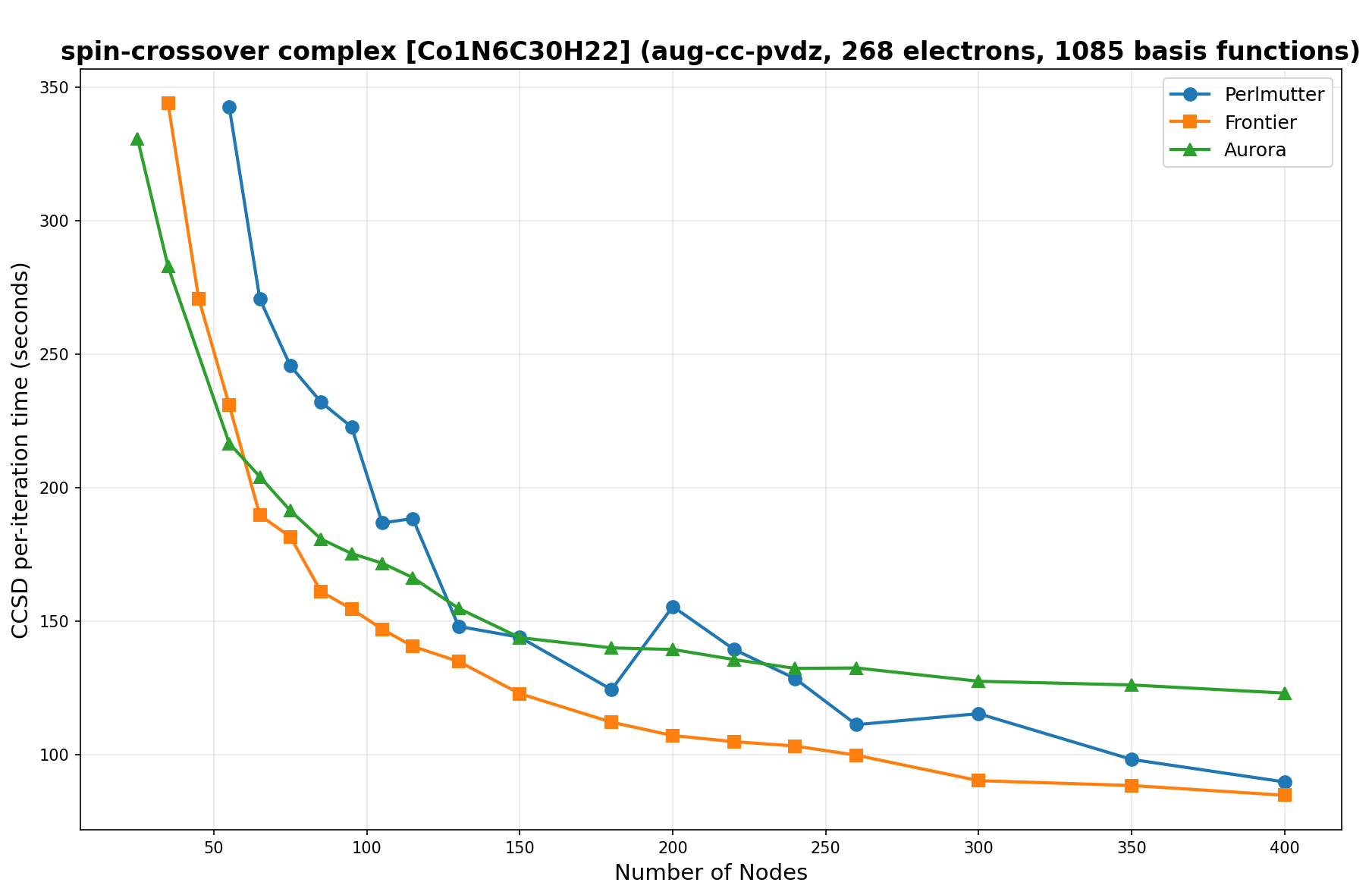
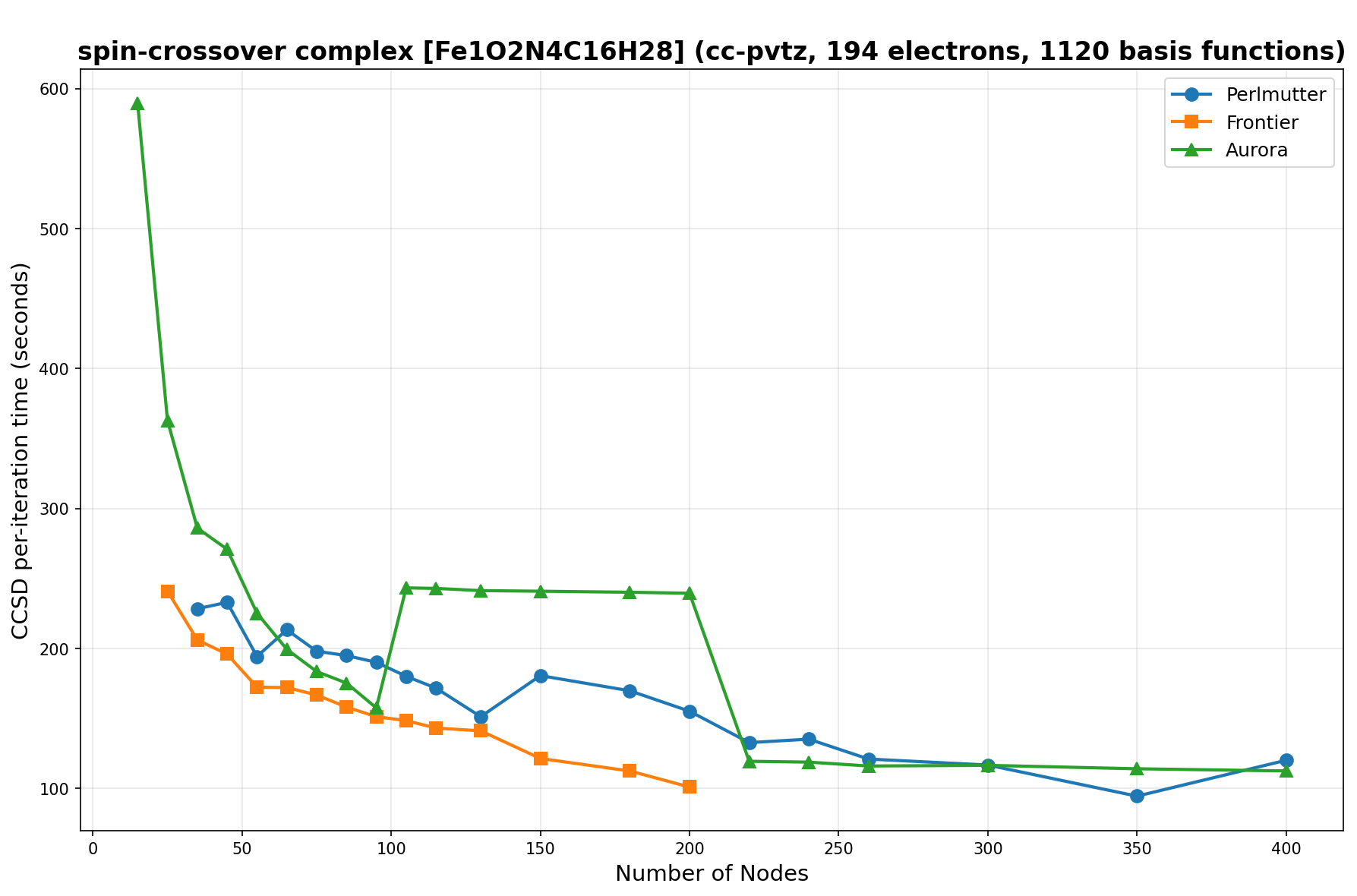
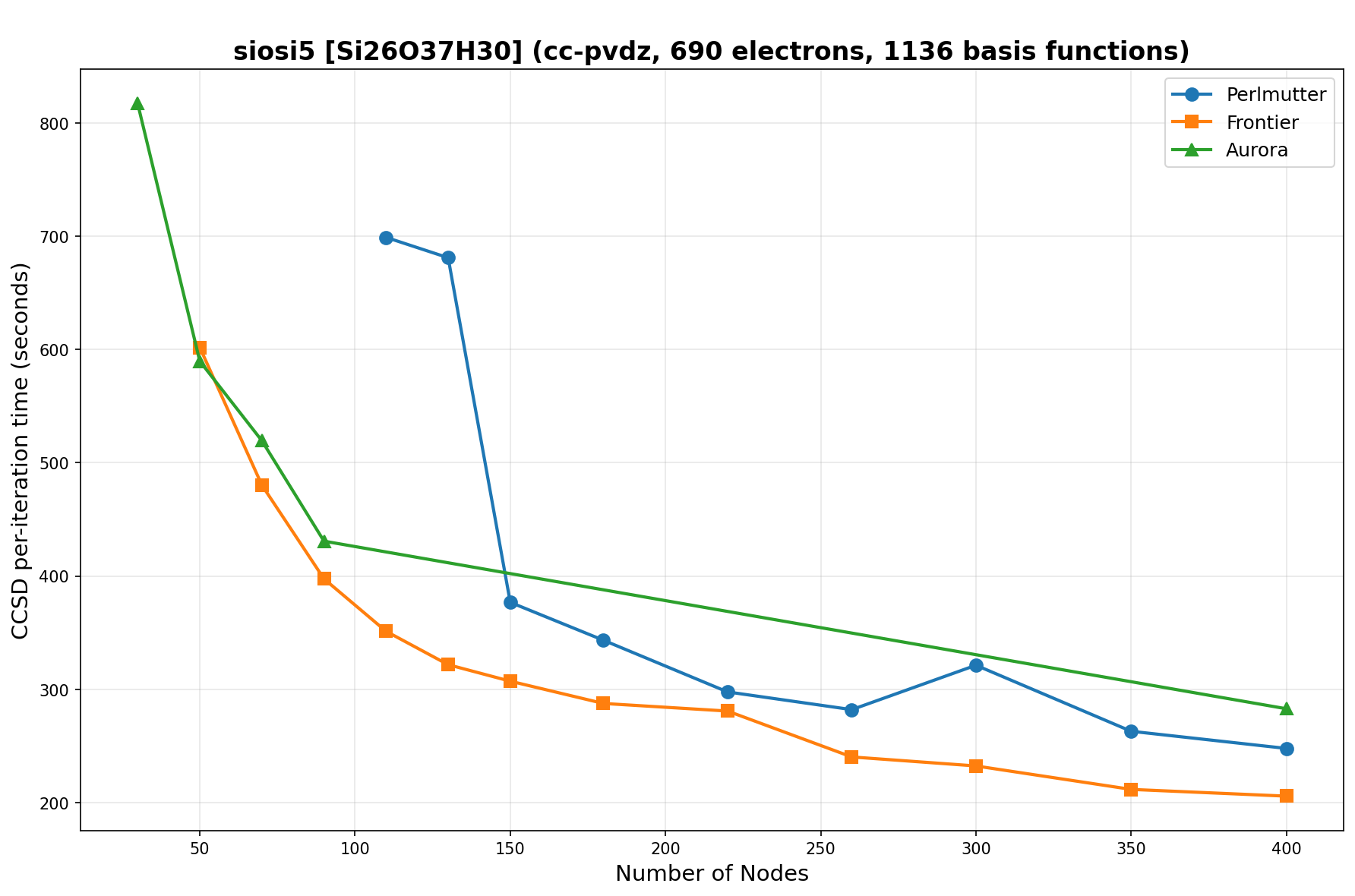
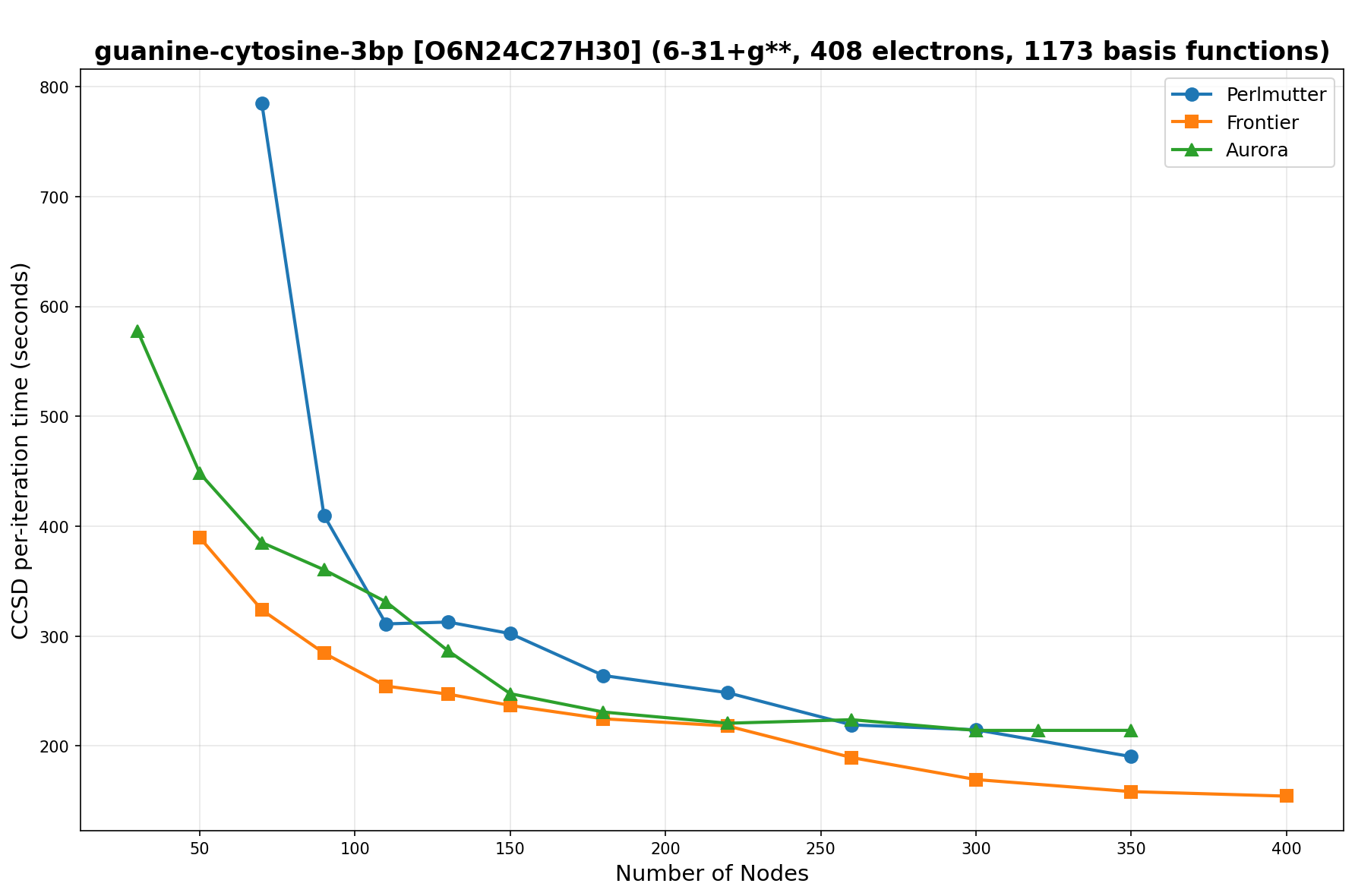
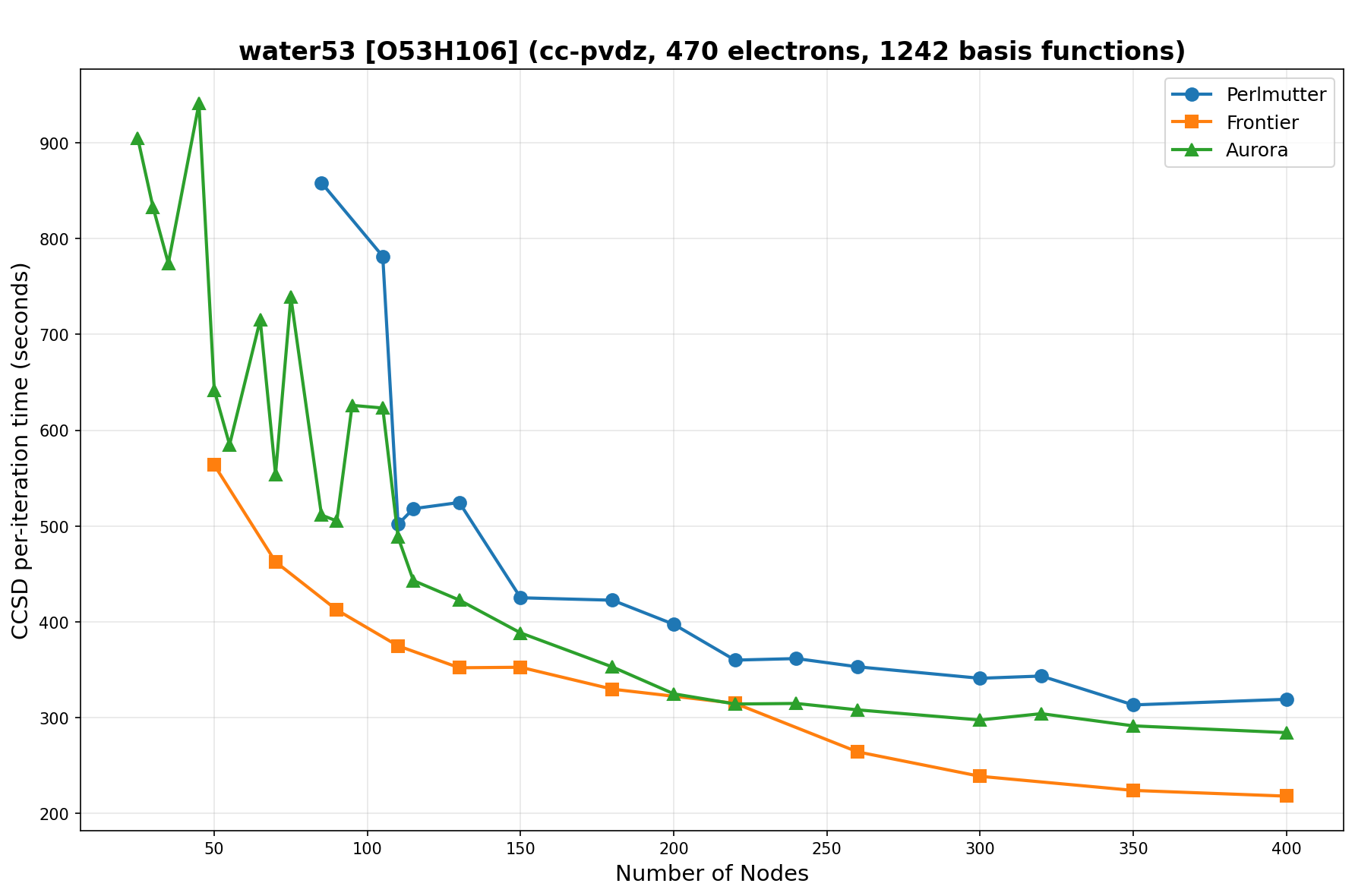
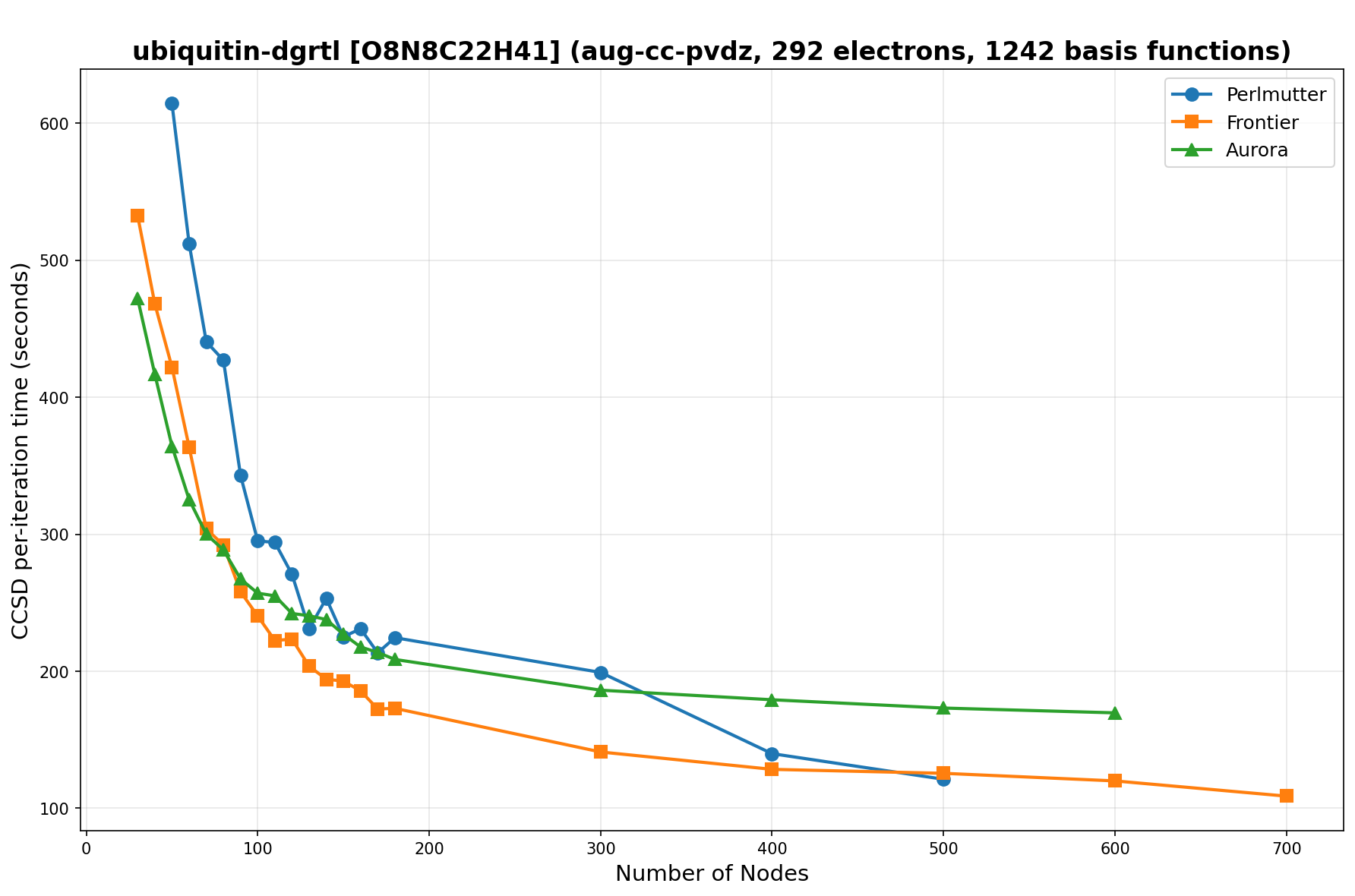
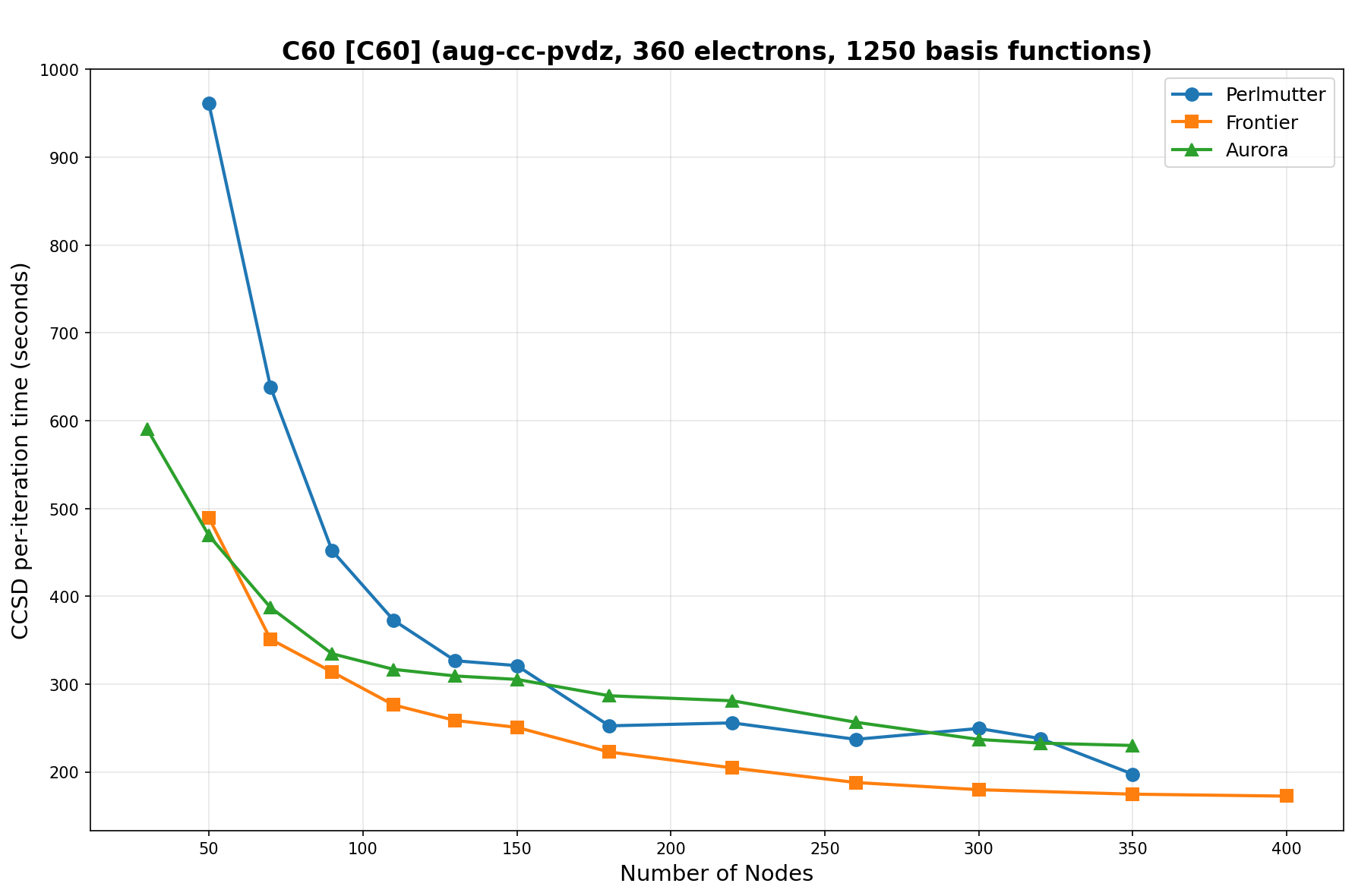
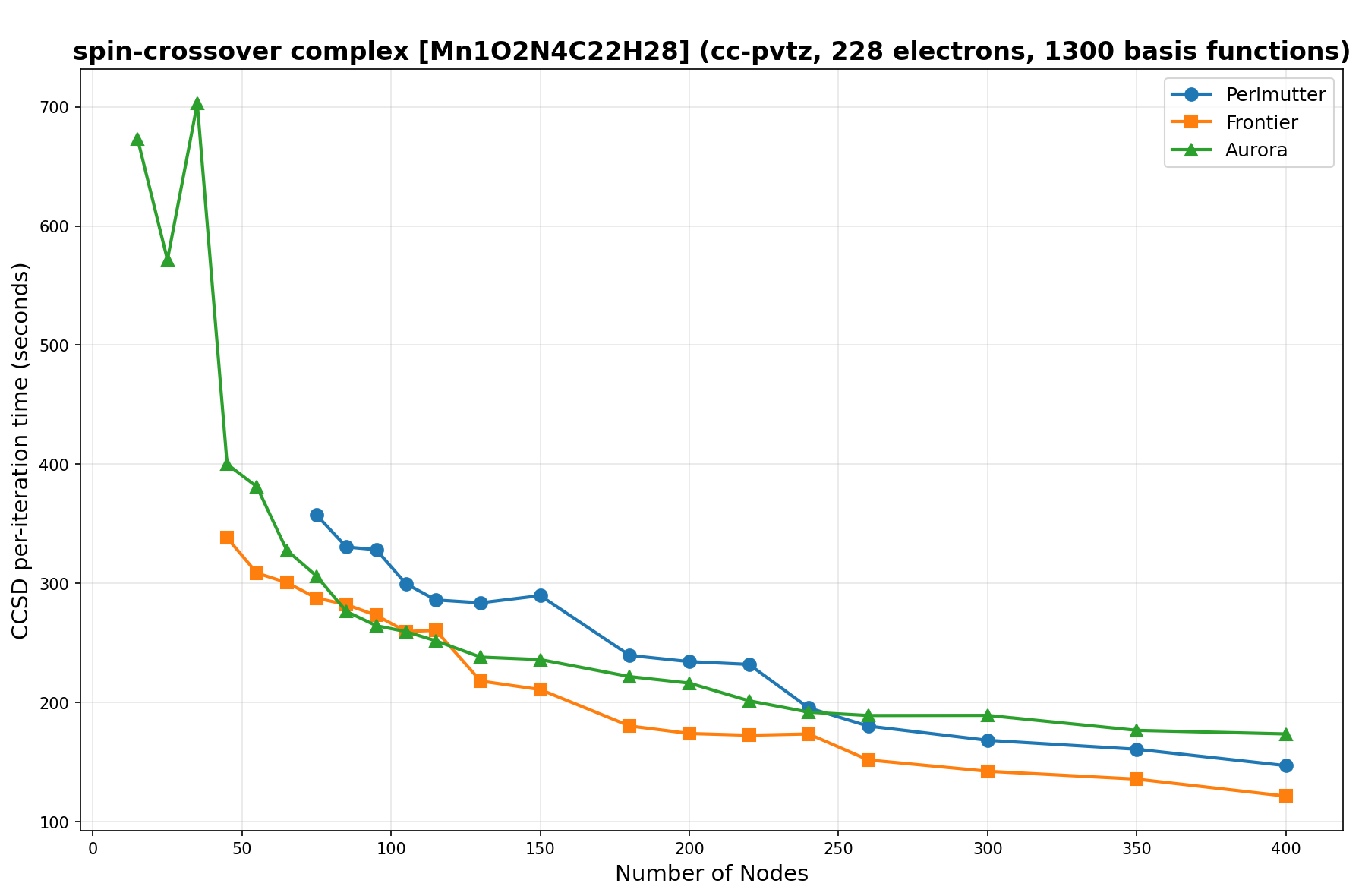
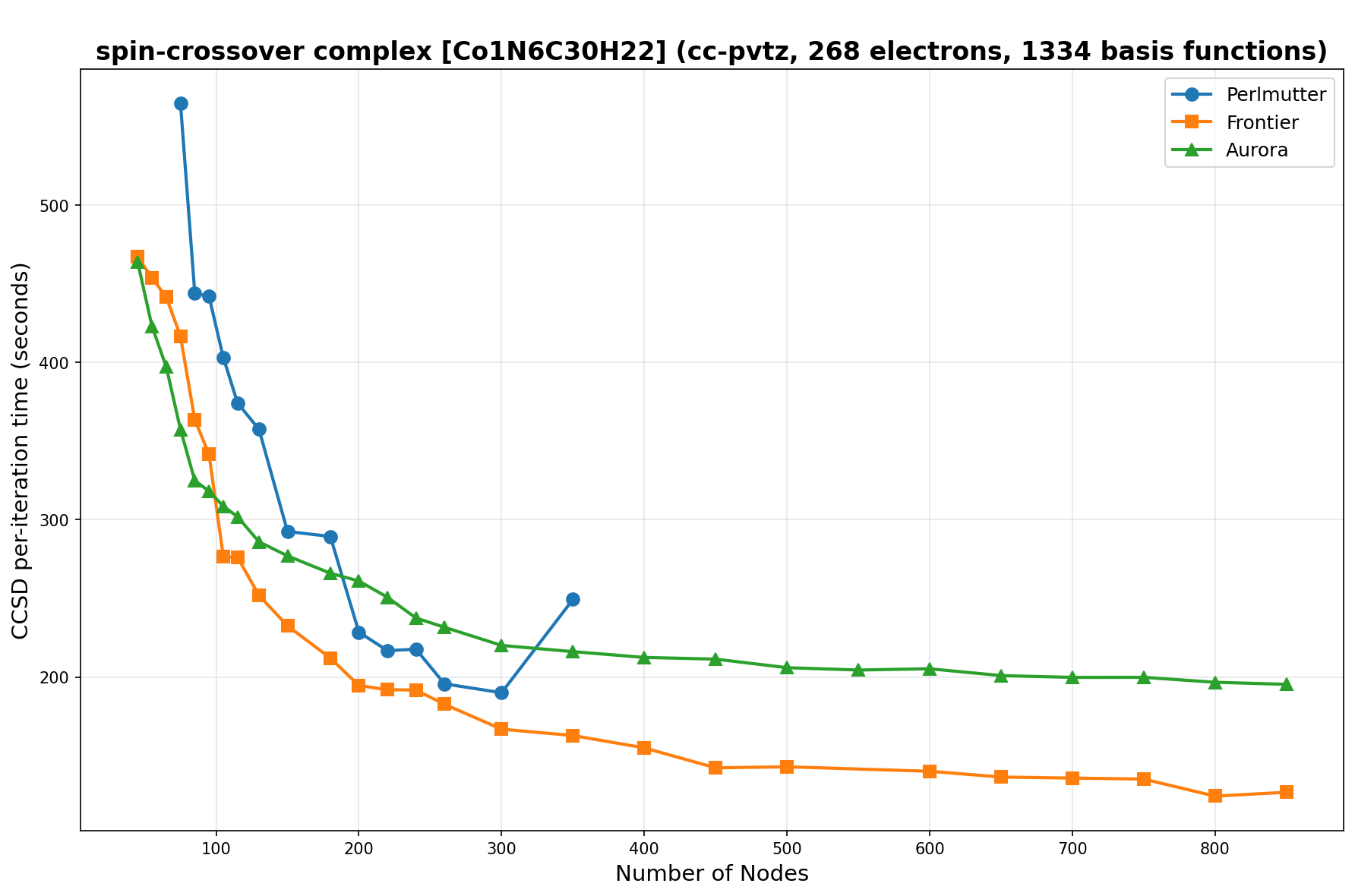
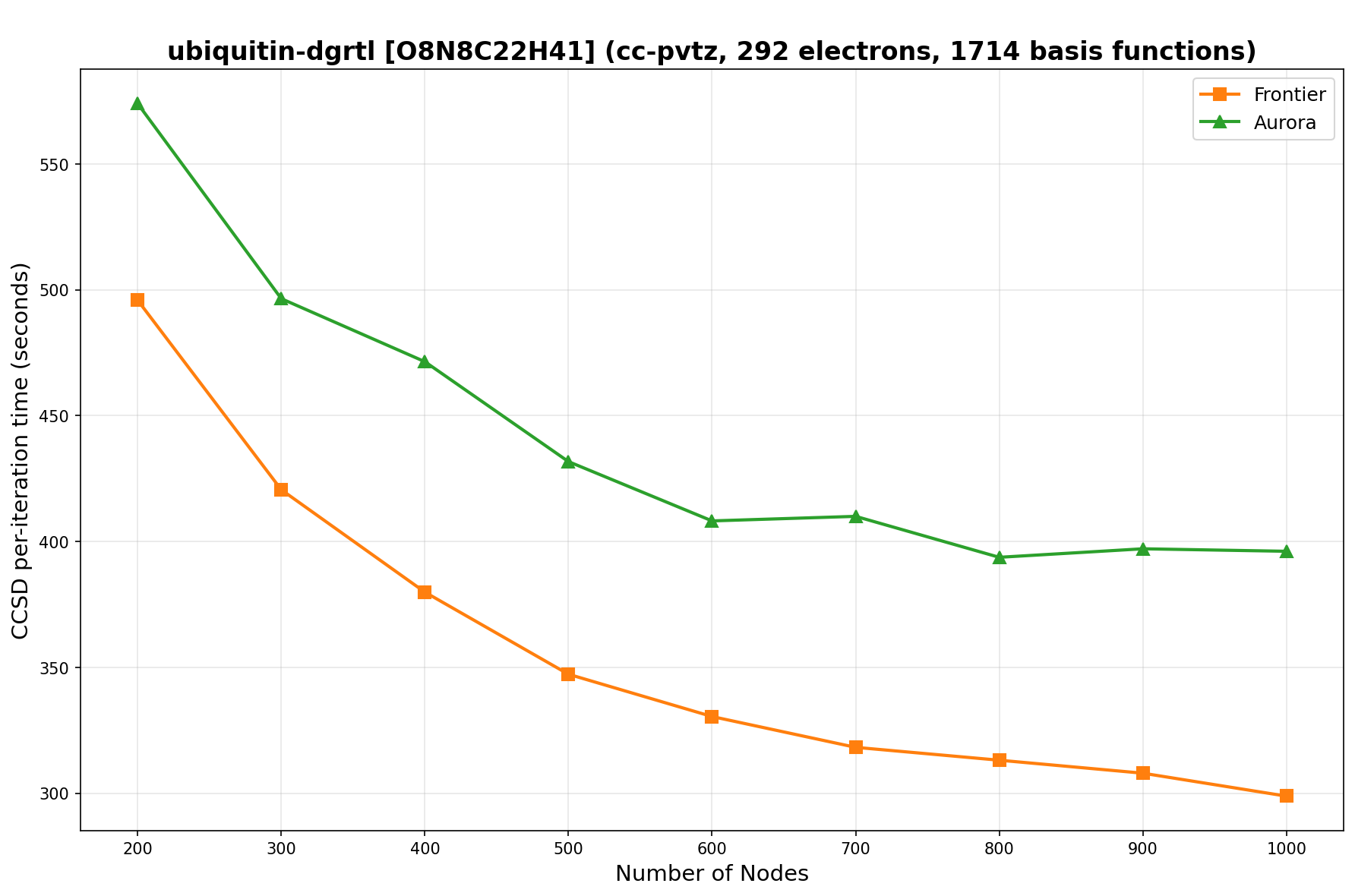
The benchmarks show the performance using the CCSD per-iteration timing on all three supercomputing platforms for a variety of molecular configurations. All benchmarks shown are closed-shell calculations and use an explicitly correlated all-electron treatment with no frozen core. The benchmarks are arranged in increasing order of basis functions. On each machine, the number of MPI processes per node is set to the number of GPU tiles per node - 4 for Perlmutter, 8 for Frontier, and 12 for Aurora. All molecular systems benchmarked demonstrate strong scaling with increasing node counts, though the optimal node count varies with the system and molecular configuration. For certain molecular systems, we also include data points beyond the strong-scaling limit, even where performance degrades, to highlight the scalability limits of the benchmarked systems. Please note that these timings were obtained in Summer 2025 and may evolve in the future.